PCzEB/Elementy stereochemii
Spis treści
ELEMENTY STEREOCHEMII
Stereochemia zajmuje się ustaleniem przestrzennej struktury cząsteczek oraz badaniem jej wpływu na reaktywność związków chemicznych. Ponieważ w stereochemii chodzi o wzajemne relacje pomiędzy obiektami w przestrzeni trójwymiarowej, wymaga ona używania pewnych pojęć rzadko spotykanych w codziennym życiu.
Chiralność – cecha obiektów trójwymiarowych (np. cząsteczek chemicznych nie posiadających płaszczyzny symetrii), przejawiająca się w tym, że cząsteczka wyjściowa i jej lustrzane odbicie nie są identyczne i nie można ich nałożyć na siebie na drodze translacji i obrotu w przestrzeni.

Takie obiekty stanowią parę enancjomerów. Ich równomolowa mieszanina zwana jest mieszaniną racemiczną lub racematem.
Warunkiem wystarczającym chiralności danego obiektu jest brak
występowania jakiegokolwiek elementu wewnętrznej symetrii (środka, osi
lub płaszczyzny symetrii), czyli jego pełna asymetria. Nie jest
to jednak warunek konieczny: istnieją bowiem obiekty chiralne
posiadające dwukrotną oś symetrii, będącą wszak elementem
symetrii. Takie obiekty nazywamy dysymetrycznymi (lecz nie można
ich nazwać asymetrycznymi). Warunkiem koniecznym i wystarczającym
chiralności jest nieposiadanie przez obiekt ani inwersyjnej osi
symetrii, ani płaszczyzny symetrii.

Czynność optyczna
Zjawisko to zaobserwowano przepuszczając przez niektóre roztwory światło liniowo spolaryzowane.
Różnica między światłem niespolaryzowanym a światłem liniowo spolaryzowanym polega na tym, że w pierwszym przypadku wektor pola elektrycznego (oraz prostopadle do niego położony wektor pola magnetycznego) położony jest we wszystkich możliwych płaszczyznach przechodzących przez oś kierunku propagacji fali a w drugim – tylko w jednej płaszczyźnie, zwaną płaszczyzną polaryzacji (zwyczajowo jest to płaszczyzna drgań pola elektrycznego, a nie magnetycznego).
.svg)
Podczas przechodzenia przez chiralny ośrodek (np. roztwór związku chiralnego, nieracemicznego), płaszczyzna światła spolaryzowanego ulega skręceniu zgodnie (+) lub przeciwnie (-) do ruchu wskazówek zegara. Dwa związki stanowiące parę enancjomerów, mają skręcalność równą co do wartości bezwzględnej, lecz z przeciwnym znakiem. Z reguły znak ten podaje się przy nazwie chiralnego związku (nawet, jeżeli jego konfiguracja absolutna nie jest znana).
Skręcalność optyczna w rzeczywistości ma związek z różną prędkością propagacji (i co za tym idzie, różnym współczynnikiem załamania) światła prawo- i lewoskrętnie kołowo spolaryzowanego.
Polaryzację liniową można bowiem uważać za superpozycję dwóch możliwych dla danej długości fali polaryzacji kołowych.
.svg)
Upraszczając - można powiedzieć, iż każda chiralna molekuła w próbce, przez którą przechodzi światło liniowo spolaryzowane, skręca płaszczyznę jego polaryzacji o pewną bardzo niewielką wartość, w „prawo” (+) lub „lewo” (-). W przypadku mieszanin racemicznych, tyle samo jest molekuł które skręcają zgodnie i przeciwnie do ruchu wskazówek zegara, dlatego skręcalność optyczna racematu zawsze równa jest 0. Skręcalność rośnie w przybliżeniu liniowo wraz ze stężeniem molowym i zmienia się w zależności od rozpuszczalnika (czasami znacznie, włącznie ze zmianą znaku skręcalności przy zmianie rozpuszczalnika). Wartości bezwzględne skręcalności optycznej z reguły rosną wraz ze zmniejszaniem się długości fali świetlnej, lecz przy pewnej długości absorpcja światła przez próbkę staje się na tyle duża, że znacząco utrudnia pomiary.
Skręcalność próbki o stężeniu 10 mg/mL, w kuwecie o długości 10 cm, dla długości fali 589 nm (linia D sodu) jest zdefiniowana jako skręcalność właściwa [α]D. Skręcalność właściwa zależy tylko od rozpuszczalnika oraz temperatury (dane te podaje się zawsze podczas podawania skręcalności właściwej danej substancji).
Istnieją także związki, które pomimo posiadania centrum stereogenicznego, nie wykazują czynności optycznej. Związki takie nazywa się kryptochiralnymi. Wśród substancji kryptochiralnych znajduje się większość tłuszczów właściwych, będących estrami glicerolu oraz długołańcuchowych kwasów karboksylowych (kwasów tłuszczowych). Jest to jednak cecha dość wyjątkowa – można przyjąć, iż nieracemiczny związek chiralny najprawdopodobniej będzie posiadał niezerową czynność optyczną.
Czynność optyczna związana jest też z innym zjawiskiem zwanym dichroizmem kołowym (CD – circular dichroizm). Jego przyczyna także leży w różnym oddziaływaniu chiralnego ośrodka ze światłem kołowo spolaryzowanym, lecz w tym wypadku chodzi nie o różnicę w prędkości propagacji, lecz o różnicę w absorpcji. Widmo CD w świetle widzialnym i ultrafiolecie daje istotne informacje na temat budowy i stereochemii badanej substancji.
W chemii przyjmujemy zgodnie z zasadą Curie (Pierre Curie, 1859-1906), iż chiralność musi być indukowana i rozróżniana przez inną chiralność. Jak widać, nawet w przypadku światła, możemy mówić o jego „chiralności” ze względu na możliwość polaryzacji kołowej i eliptycznej.
Tylko dlatego światło płasko spolaryzowane może „rozróżniać” enancjomery.
IZOMERY
Izomeria to relacja, wiążąca cząsteczki o takim samym składzie atomowym, lecz różnej budowie przestrzennej. Dlategoteż izomery są związkami chemicznymi o identycznych sumarycznych wzorach cząsteczkowych, różniące się między sobą sposobem połączenia atomów, ich kolejnością, albo ich innym rozmieszczeniem w przestrzeni.

Izomery konstytucyjne
różnią się kolejnością połączenia atomów w cząsteczce.
| C2H6O | C2H6O |
| CH3CH2–OH | CH3–O–CH3 |
| alkohol etyowy | eter dimetylowy |
Izomery konformacyjne
różnią się wzajemnym ułożeniem atomów w przestrzeni, konformery mogą się wzajemnie w siebie przekształcać, na przykład na skutek obrotu wokół wiązania pojedynczego.


Izomery konfiguracyjne (stereoizomery)
różnią się rozmieszczeniem atomów w przestrzeni i w przeciwieństwie do izomerów konformacyjnych nie przekształcają się w siebie wzajemnie.
Enancjomery
są stereoizomerami, których cząsteczki mają się do siebie jak obiekt i jego lustrzane odbicie. Charakterystyczną cechą enancjomerów jest chiralność molekuł. W najprostszym przypadku enancjomery posiadają jedno centrum stereogeniczne (niepoprawnie zwane także „centrum chiralnym”. Pamiętać należy, iż chiralność jest cechą całego obiektu, nie dotyczy więc pojedynczych atomów, lecz cząsteczek). Najczęściej centrum stereogeniczne stanowi atom węgla podstawiony czterema różnymi grupami, ale nie jest to jedyna możliwość. Mogą też istnieć enancjomery, które nie posiadają centrum stereogenicznego zlokalizowanego na jakimkolwiek z atomów, albowiem elementem generującym enancjomerię (stereogennym) jest oś, płaszczyzna lub cała przestrzeń (spirala).
Centrów stereogenicznych może być wiele, przy czym dla pary enancjomerów konfiguracje na wszystkich centrach muszą być przeciwne (tylko wtedy mamy do czynienia z „lustrzanymi odbiciami”).
Parę enancjomerów stanowi zawsze para związków, które mają przeciwne konfiguracje na wszystkich centrach stereogenicznych.
Równomolową mieszaninę enancjomerów nazywamy mieszaniną racemiczną lub racematem.
Enancjomery mają identyczne widma NMR. Z reguły widma czystych enancjomerów są też identyczne z widmami NMR racematu.
Większość właściwości fizycznych substancji, które stanowią parę enancjomerów jest identyczna, poza skręcalnością optyczną, oraz dichroizmem kołowym.
Diastereoizomery (diastereomery)
— stereoizomery, które są nienakładalne na siebie i nie są swoim odbiciem lustrzanym. Zaliczamy do nich:
- izomery geometryczne (izomeria spowodowana zahamowaną rotacją wokół wiązań, zarówno podwójnych, jak i w pierścieniach).
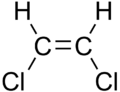 120px
120px
cis-1,2-dichloroeten i trans-1,2-dichloroeten
- diastereomery zawierające minimum 2 centra stereogeniczne, przy czym których przynajmniej jedno ma tę samą konfigurację a co najmniej jedno – przeciwną. Związki będące diastereoizomerami posiadają w ogromnej większości przypadków różne widma NMR.


L-allo-Treonina (2S,3S) i D-allo-Treonina (2R,3R)



Początki stereochemii
Odkrycie enancjomerii wśród molekuł i tym samym stworzenie podstaw stereochemii, jest zasługą Ludwika Pasteura. Krystalizował on między innymi sole kwasu winowego, substancji naturalnej powstającej podczas fermentacji winnej. Podczas rekrystalizacji winianu sodowo-amonowego z roztworu wodnego, w temperaturze zbliżonej do pokojowej zauważył w otrzymanym osadzie dwa rodzaje bardzo podobnych do siebie kryształów. Różniły się jedynie tak, jak różni się przedmiot od swojego odbicia w lustrze (enancjomorficzność).
Pasteur doszedł do wniosku, że różnice wyglądu zewnętrznego kryształu wynikają z różnic przestrzennej budowy cząsteczki. Inaczej mówiąc, niektóre cząsteczki kwasu winowego stanowią odbicie lustrzane innych — są w stosunku do siebie enancjomerami.
Pasteur rozdzielił te kryształy za pomocą pęsety i stwierdził, że ich skład chemiczny, a także właściwości fizykochemiczne były identyczne. Różniły się jedynie znakiem skręcalności właściwej. Roztwór o tym samym stężeniu sporządzony z kryształów jednego rodzaju skręcał światło spolaryzowane w polarymetrze o ten sam kąt lecz w przeciwną stronę, niż roztwór sporządzony z kryształów drugiego rodzaju. Jedna odmiana winianu sodowo-amonowego skręcała zatem w prawo (+), a druga w lewo (-).
Zostały one nazwane prawoskrętną i lewoskrętną odmianą tego związku. Obie odmiany zmieszane razem traciły zdolność skręcania płaszczyzny światła spolaryzowanego, podobnie jak przed rozdzieleniem stawały się optycznie nieczynne. Taka mieszanina została nazwana mieszaniną racemiczną. Dzięki innemu przestrzennemu ułożeniu podstawników przy C2 i C3 kwasu winowego powstają trzy jego odmiany: lewoskrętna (-), prawoskrętna (+) i optycznie nieczynna zwana kwasem mezo-winowym (cząsteczka posiada płaszczyznę symetrii).
- Izomery optyczne kwasu winowego
|
|

|

|
|
kwas (+)-winowy |
kwas (-)-winowy |
kwas mezo-winowy |

Symbole (R) i (S) oznaczają tzw. konfigurację absolutną oznaczoną, określoną wg reguły Cahna-Ingolda-Preloga:
- ustala się kolejność pierwszeństwa podstawników przyłączonych do atomu stanowiącego centrum stereogeniczne
- podstawnik o najniższym pierwszeństwie (D) musi znajdować się jak najdalej od obserwatora
- jeśli układ pozostałych podstawników jest taki, że patrząc od strony obserwatora należy wodzić okiem od podstawnika o największym pierwszeństwie (A) do trzeciego w kolejności (C) zgodnie z kierunkiem wskazówek zegara to konfiguracja absolutna jest oznaczana literą R (do łac. rectus — prawy) , a gdy odwrotnie literą S (od łac. sinister — lewy).
 (S)-(+)-kwas mlekowy (lewy) and (R)-(–)-kwas mlekowy (prawy) są swoimi lustrzanymi odbiciami
(S)-(+)-kwas mlekowy (lewy) and (R)-(–)-kwas mlekowy (prawy) są swoimi lustrzanymi odbiciami

{kind=link}
{kind=link}
W ramach konwencji Cahna-Ingolda-Preloga istnieją cztery reguły ustalania kolejności podstawników:
- liczba atomowa (l.a) atomu podstawnika, który jest bezpośrednio przyłączony do centrum chiralności, np.: w sytuacji, gdy podstawniki są czterema różnymi pojedynczymi atomami (np.: jod (I, l.a = 53), brom (Br, l.a = 35), chlor (Cl, l.a= 17), wodór (H, l.a = 1) ich kolejność jest następująca: I > Br > Cl > H;
- w wypadku występowania izotopów, o pierwszeństwie decyduje ich większa liczba masowa;
- jeśli podstawniki łączą się z centrum chiralności tymi samymi atomami, to bierze się pod uwagę kolejne atomy przyłączone bezpośrednio do atomu, którym cały podstawnik łączy się z centrum chiralności;
- gdy dalsze atomy są połączone wiązaniami wielokrotnymi, liczy się je jakby były połączone wiązaniami pojedynczymi, tyle że wiele razy.
Należy podkreślić, że nie istnieje żadna prosta zależność pomiędzy konfiguracją absolutną na centrum, lub centrach stereogenicznych a znakiem skręcalności światła liniowo spolaryzowanego. Dlatego podaje się w osobnych nawiasach konfigurację absolutną i znak skręcalności. Możemy być natomiast pewni, że drugi enancjomer danego związku będzie miał skręcalność równą co do wartości, lecz o przeciwnym znaku.
Chiralne kryształy
Ciekawym wnioskiem płynącym z rozważań na temat substancji enancjomerycznie czystych, jest to, iż nie mogą one krystalizować z utworzeniem kryształów centrosymetrycznych. Oznacza to, iż komórka elementarna takich kryształów, podobnie jak i całe kryształy, nie mogą nigdy posiadać środka symetrii.
Związki chiralne, na przykład dwutlenek krzemu, także mogą (lecz nie muszą!) tworzyć kryształy niecentrosymetryczne (w wypadku SiO2 — kwarc).
Z pozoru nieistotna cecha, jaką jest niecentrosymetryczność kryształów, niesie za sobą ważne konsekwencje — kryształy takie mogą posiadać pewne unikalne właściwości nigdy nie występujące w kryształach centrosymetrycznych. Należą do nich:
- podwójne załamanie (dwójłomność) — podczas przechodzenia przez kryształ, promień świetlny ulega rozdzieleniu na tzw. promień zwyczajny i nadzwyczajny. Podwójne załamanie jest szeroko wykorzystywane w wielu dziedzinach optyki.
- piezoelektryczność — pod wpływem sił mechanicznych działających na kryształ, dochodzi do separacji ładunków elektrycznych i powstania potencjału. Jednocześnie przyłożenie napięcia do kryształu, wywołuje jego drgania mechaniczne.
- tryboluminescencja emisyjna — podczas łamania kryształu dochodzi do emisji kwantów promieniowania elektromagnetycznego.
Kryształy związków czystych enancjomerycznie (czystych optycznie) dają możliwość eksperymentalnego zmierzenia ich konfiguracji absolutnej. Rentgenowska analiza strukturalna monokrystalicznej substancji nie pozostawia żadnych wątpliwości.
Rentgenowska analiza strukturalna
Czy można uzyskać „zdjęcie” cząsteczki chemicznej? Tak! Choć porównanie do fotografii jest nieco zbyt daleko idącym uproszczeniem, to dzięki rentgenowskiej analizie strukturalnej jesteśmy w stanie uzyskać trójwymiarowy obraz komórki elementarnej, z uwzględnieniem przestrzennego ułożenia atomów — a więc i rzeczywisty przestrzenny obraz pojedynczej molekuły.
Rentgenografia strukturalna jest techniką analityczną, opartą na rejestracji obrazów dyfrakcyjnych promieniowania X (rentgenowskiego), przechodzącego przez kryształ pod różnymi kątami. Wysokoenergetyczne kwanty promieniowania, przechodząc przez kryształ oddziałują z chmurami elektronowymi atomów, tworząc trójwymiarową mapę gęstości elektronowej. Matematyczna obróbka obrazu dyfrakcyjnego umożliwia:
- wyznaczenie pozycji i odległości cząsteczek względem siebie w sieci krystalicznej,
- wyznaczenie położenia poszczególnych atomów względem siebie,
- ustalenie kątów i długości wiązań między atomami,
- ustalenie rozkładu gęstości chmur elektronowych wokół poszczególnych atomów, co umożliwia obliczenie momentu dipolowego wiązań i całych cząsteczek oraz precyzyjne ustalenie natury poszczególnych wiązań.
Dzięki olbrzymiemu postępowi w tej dziedzinie analitycznej, obecnie możliwe jest wykonywanie rentgenostruktur nawet olbrzymich białek i innych makromolekuł.
Ogromną zaletą tej metody jest możliwość dokładnego ustalenia struktury chemicznej związków chemicznych z niemal absolutną pewnością, umożliwiającą zbudowanie ich rzeczywistego modelu przestrzennego. Żadna inna metoda analityczna nie daje takiej pewności i zawsze zostawia możliwość różnej interpretacji wyników.
Wadą rentgenografii jest konieczność uzyskania czystego monokryształu analizowanego związku chemicznego o wymiarach liniowych rzędu 0,1mm. W przypadku niektórych związków chemicznych wyhodowanie takiego kryształu jest z wielu względów bardzo trudne, a czasem wręcz niemożliwe. Nierzadko krystalizacja białek trwa latami i jest wielkim osiągnięciem badawczym. Inną wadą rentgenografii jest stosunkowo wysoki koszt i czasochłonność wykonywania takiej analizy, wynikające także z trudności manualnych związanych z operowaniem bardzo niewielkim kryształem. Powoduje to, iż przy wykonywaniu analizy liczy się gruntowne wyszkolenie i doświadczenie operatora.
Nowoczesny monokrystaliczny dyfraktometr rentgenowski kosztuje w granicach 100—500 tys.€. Pomiar danych dla przeciętnego związku organicznego lub metaloorganicznego zabiera w zależności od urządzenia od kilku godzin do ok. dwóch tygodni. Analiza otrzymanych danych (rozwiązanie struktury związku), jeśli jego struktura jest w miarę prosta, jest dość szybka, natomiast w przypadku bardzo złożonych struktur, np. kryształów białek, czas ten może wynosić nawet kilka tygodni.
Spektroskopia NMR
— od ang. Nuclear Magnetic Resonance — Spektroskopia Jądrowego Rezonansu Magnetycznego, to obok analizy rentgenowskiej, jedna z najważniejszych technik analitycznych w chemii, oraz diagnostycznych w medycynie (MRI — Magnetic Resonance Imaging).
Spektroskopia NMR polega na wzbudzaniu spinów jąder, znajdujących się w silnym zewnętrznym polu magnetycznym, poprzez szybkie zmiany pola magnetycznego, a następnie rejestrację promieniowania elektromagnetycznego powstającego na skutek zjawisk relaksacji, gdzie przez relaksację rozumiemy powrót układu spinów jądrowych do stanu równowagi termodynamicznej. NMR jest zatem jedną ze spektroskopii rezonansowych.
Ważnym ograniczeniem jest fakt, iż łatwo możemy obserwować tylko jądra o spinie niecałkowitym. 1H, 13C, 15N, 19F i 31P są jądrami o największym znaczeniu w spektroskopii NMR:
- 1H z uwagi na dużą czułość i występowanie w licznych związkach chemicznych,
- 13C ze względu na to, że węgiel jest głównym składnikiem związków organicznych (mimo że 13C ma niewielką zawartość w stosunku do izotopu 12C, który ma spin 0 i jest nieaktywny w NMR),
- 15N z uwagi na występowanie azotu w kluczowych w biochemii związkach: białkach i DNA (mimo iż 15N ma znikomą zawartość w stosunku do izotopu 14N, który ma niezerowy moment kwadrupolowy, co powoduje poszerzenie sygnałów NMR),
- 19F z uwagi na dużą czułość,
- 31P z uwagi na częste wstępowanie z związkach organicznych (w tym DNA) i dość dużą czułość.
Spektroskopia NMR ta jest w chemii organicznej podstawową techniką analityczną. W odróżnieniu od spektrometrii mas, daje ona bardzo dużo informacji na temat budowy przestrzennej związku chemicznego. Więcej informacji daje tylko rentgenowska analiza strukturalna, jednak tam potrzebne są związki w postaci monokryształów, a takie często nie dadzą się uzyskać, lub ich uzyskanie jest bardzo czasochłonne.
W tomografii MRI wykorzystuje się rezonans magnetyczny protonów zawartych w molekułach wody, uzyskując z dużą dokładnością obraz jej zawartości w poszczególnych narządach. Ponieważ różne tkanki i narządy zawierają różne proporcje wody w stosunku do innych składników, możliwe jest uzyskanie dokładnych przekrojów organizmu, w sposób całkowicie nieinwazyjny.
W chemii organicznej zaletą spektroskopii NMR jest to, iż próbka może się znajdować w roztworze. Ponieważ większość rozpuszczalników zawiera atom wodoru, w praktyce należy wżywać rozpuszczalników deuterowanych — takich, w których udział izotopu 2H został zwiększony z ok. 1% do ponad 99.5%. Rozpuszczalnika jest zwykle (molowo) znacznie więcej niż próbki, więc jądra 1H (protu) całkowicie „zakrywałyby” sygnały od próbki.
Dzięki ekranowaniu badanego jądra przez inne jądra w badanej molekule (zwykle — co bardzo istotne — nie obserwuje się oddziaływań pomiędzy dwoma molekułami; dzięki temu stężenie próbki jest mało istotne), a także dzięki ekranowaniu przez elektrony jądra o różnym otoczeniu chemicznym dają różne sygnały na widmie NMR. To właśnie dzięki temu ilość informacji, jaką można uzyskać dzięki opisywanej technice jest tak duża.
Bez wnikania w szczegóły, poprawnie wykonane widmo NMR daje informacje na temat:
- obecności charakterystycznych grup funkcyjnych w badanej molekule organicznej,
- wzajemnego położenia danych jąder — stereochemii molekuły,
- w przypadku 1H—NMR — względny stosunek ilościowy nieidentycznych chemicznie jąder,
- niektórych procesów chemicznych zachodzących w badanej próbce.
Spośród możliwych izomerów, teoretycznie tylko enancjomery posiadają identyczne widma NMR. Dlatego bezpośrednio nie jesteśmy w stanie powiedzieć czy w skład próbki wchodzi tylko jeden enancjomer, czy któryś z nich jest w przewadze, czy też jest to mieszanina racemiczna (patrz rodział Stereochemia). I tutaj istnieje jednak pewien sposób — dodanie do próbki enancjomerycznie czystych związku, mogącego kompleksować badany produkt powoduje powstawanie dwóch diastereomerycznych kompleksów, które mogą być już rozróżnione. Jak zawsze działa tu zasada Curie — tylko chiralność może rozróżniać drugą chiralność (patrz wyżej).
Dzięki powyższym cechom spektroskopii NMR, dobry chemik — przy wykorzystaniu danych widmowych oraz wiedzy chemicznej — bardzo często jest w stanie poznać nawet subtelne szczegóły strukturalne badanego związku, a także wnioskować o zmianach jego budowy podczas przebiegu reakcji chemicznych.
Znaczenie poznania struktury związków chemicznych
Znaczenie znajomości dokładnej budowy związku, łącznie z określeniem konfiguracji absolutnej na wszystkich centrach stereogenicznych występujących w cząsteczce jest ogromne, ze względu na to iż podstawowe „cegiełki” budulcowe organizmów żywych są chiralne, i enancjomerycznie czyste (na przykład praktycznie wszystkie aminokwasy białkowe poza L—cysteiną, mają tę samą konfigurację absolutną S. R-aminokwasy rzadko kiedy wchodzą w skład organizmów! Związki chemiczne występujące w organizmach żywych i w konsekwencji struktury biologiczne z nich zbudowane, precyzyjnie „rozpoznają” (różnicują) enancjomery. Skoro receptory, odczytujące informację chemiczną niesioną przez molekuły substancji leczniczej są chiralne, to oba enancjomery danego leku będą miały różną aktywność metaboliczną.
Dobrze obrazuje to następujący, smutny przykład
Talidomid — lek działający przeciwwymiotnie, przeciwbólowo, nasennie i uspokajająco — otrzymany był jako mieszanina racemiczna, tzn. zawierająca w równych proporcjach oba enancjomery. Okazało się niestety, że jeden z enancjomerów (o konfiguracji absolutnej R) ma działanie lecznicze, a drugi (S) jest silnym mutagenem zaburzającym embriogenezę. Tragedia polegała m.in. na tym, iż talidomid był wprowadzony do lecznictwa jako lek uspokajający przeznaczony dla kobiet w ciąży.
  |
| (S)-talidomid i (R)-talidomid |
Zanim nagłośniono teratogenne działanie tego związku, jego ofiarami zostało ok. 15 000 ciężarnych kobiet, z czego 12 000 ciąż zostało donoszonych, a urodzone dzieci miały głębokie wady wrodzone. Pierwszy rok życia przeżyło ok. 8 000 dzieci. Większość z ocalałych dzieci żyje do tej pory, choć niemal wszystkie mają ciężkie deformacje ciała, na które składają się przedwszystkim brak kończyn i bardzo nienaturalne proporcje. Po tych odkryciach talidomid został wycofany z lecznictwa i wstrzymano na wiele lat jego produkcję. Naukowcy zbadali, który z enancjomerów ma działanie lecznicze, a który jest odpowiedzialny za działanie mutagenne, a następnie opracowali syntezę asymetryczną prowadzącą do otrzymania tylko enancjomery o działaniu leczniczym. Okazało się jednak, że w organizmach żywych związek ten ulega stopniowej racemizacji, a zatem nawet podanie czystego izomeru o działaniu leczniczym nie jest do końca bezpieczne.